Creating a workflow¶
In bioinformatics, a pipeline is defined as a sequence of bioinformatics tools that transform and analyze the input data. A pipeline in CloudConductor is representes as a directed graph of modules, where a module is a bioinformatics tool. Consider Figure 1 as an example of a pipeline.
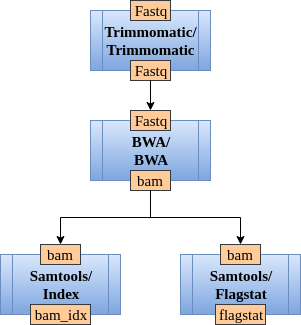
Before we explain how to define a pipeline, let’s first describe how to define a module in a pipeline.
Modules¶
A module is a bioinformatics tool and a submodule is a function that a bioinformatics tool is performing.
Some bioinformatics tools have only one function, thus their modules have only one submodule.
In Figure 1, Samtools is a module with two submodules: Index and Flagstat, while BWA is one module with one submodule with the same name.
Modules and submodules are predefined. Here is a list of currently available modules and submodules.
Please read the advanced topics if you would like to define your own modules and submodules
In a pipeline, a pipeline step is defined using the keywords module and submodule as following:
[*unique_name_of_pipeline_step*]
module=*name_of_module*
submodule=*name_of_submodule_used*
Each submodule is defined by a set of input keys, output keys and a command.
The submodule is running the command on the input keys and generates output as output_keys.
In Figure 1, submodule Index from module Samtools has
one input key (“bam” - the input BAM file) and one output key (“bam_idx” - the output BAM index file).
You can specify to keep an output file generated by a module using the keyword final_output in the module definition.
For example, if you want to keep the indexed file after running samtools index you would define the module as following:
[bam_indexing]
module=Samtools
submodule=Index
final_output=bam_idx
Additionally, using the keyword docker_image you are able to specify which Docker image from resource kit you want CloudConductor to use. If no docker_image is specified, then the tool executable is obtain from the external resources list from resource kit.
More information about resources and Docker will be presented in the definition of the resource kit.
Create a pipeline graph¶
To create a pipeline graph, you need to connect the modules using the keyword input_from. For example, in Figure 1, BWA receives the input from Trimmomatic.
The value of an input_from key is a set of defined unique pipeline steps. The pipeline presented in Figure 1 can be represented as following:
[trim_reads]
module=Trimmomatic
docker_image=Trimmomatic_docker
[align_reads]
module=BWA
input_from=trim_reads
final_output=bam
[bam_indexing]
module=Samtools
submodule=Index
docker_image=Samtools_docker
input_from=align_reads
final_output=bam_idx
[bam_summary]
module=Samtools
submodule=Flagstat
docker_image=Samtools_docker
input_from=align_reads
final_output=flagstat
As you can observe, there is no need to specify the submodules for Trimmomatic or BWA as they have only one submodule with the same name.
Also, we decided to not keep the output of Trimmomatic, but you can always add fastq as final_output to keep it.
Available modules¶
Here is a list of currently implemented modules and their submodules:
| Type | Module | Submodule |
|---|---|---|
| Tool (Module) | Annovar | Annovar |
| Bcftools | BcftoolsIndex | |
| BcftoolsNorm | ||
| Bowtie2 | Bowtie2 | |
| BwaAligner | BwaAligner | |
| CellRanger | CellRanger | |
| CNVkit | CNVkit | |
| Cufflinks | Cuffquant | |
| Cutadapt | Cutadapt | |
| Delly | Delly | |
| Diamond | TaxonClassify | |
| BLASTX | ||
| FastQC | FastQC | |
| GATK | HaplotypeCaller | |
| PrintReads | ||
| BaseRecalibrator | ||
| IndexVCF | ||
| FilterMutectCalls | ||
| CollectReadCounts | ||
| BedToIntervalList | ||
| Gistic2 | Gistic2 | |
| Mosdepth | Mosdepth | |
| NovoBreak | NovoBreak | |
| Picard | MarkDuplicates | |
| CollectInsertSizeMetrics | ||
| SamToFastq | ||
| SortGVCF | ||
| QCParser | FastQC | |
| PicardInsertSizeMetrics | ||
| FastQC | ||
| SamtoolsDepth | ||
| SamtoolsFlagstat | ||
| SamtoolsIdxstats | ||
| Trimmomatic | ||
| PrintTable | ||
| MosdepthDist | ||
| GATKCollectReadCount | ||
| QCReportReader | GetNumReadsFastQC | |
| GetNumReadsTrimmomatic | ||
| RSEM | RSEM | |
| Samtools | Index | |
| Flagstat | ||
| Idxstats | ||
| View | ||
| Depth | ||
| SnpEff | SnpEff | |
| SnpSiftAnnotate | ||
| SnpSiftFilter | ||
| Star | Star | |
| Trimmomatic | Trimmomatic | |
| Utils | ConcatFastq | |
| RecodeVCF | ||
| SummarizeVCF | ||
| ViralFilter | ||
| IndexVCF | ||
| BGZip | ||
| GetReadGroup | ||
| CombineExpressionWithMetadata | ||
| GetVCFChroms | ||
| GetRefChroms | ||
| Splitters | BamSplitter | BamSplitter |
| FastqSplitter | FastqSplitter | |
| RefSplitter | RefSplitter | |
| SampleSplitter | SampleSplitter | |
| TumorNormalSplitter | ||
| VCFSplitter | VCFSplitter | |
| Mergers | CNVMergers | MakeCNVPoN |
| CNVkitExport | ||
| Gatherers | GatherBams | |
| GatherVCFs | ||
| GatherGVCFs | ||
| GATKMergers | GenotypeGVCFs | |
| Mutect2 | ||
| MergeBQSRs | ||
| CatVariants | ||
| CombineGVCF | ||
| MergeBams | MergeBams | |
| MergeRNAseq | AggregateRawReadCounts | |
| AggreagateRSEMResults | ||
| Cuffnorm | ||
| QCReportMerger | Rbind | |
| Cbind | ||
| VCFMergers | VCFMerger | |
| BGZipVCFMerger | ||
| RecodedVCFMerger | ||
| VCFSummaryMerger |